

-
Product Development
-
Clinical Trials
-
Quality
-
GxP Audits
-
Regulatory Affairs
-
Pre-authorisation
- Regulatory Roadmap
- Scientific Advice
-
Product information
-
Dossier preparation
- Dossier Gap Analysis
- MA Handling
- Post-authorisation
- Regulatory outsourcing
-
Pre-authorisation
-
Pharmacovigilance
-
QP Service / Importer
Data submission on authorised medicines (Article 57) – XEVMPD Management
Why do Marketing Authorisation Holders need to maintain the Article 57 database?
All companies that hold marketing authorisations for medicines in the European Union (EU) and the European Economic Area (EEA) are legally required to provide the European Medicines Agency (EMA) with details about their authorised medicines and ensure this information remains current. This obligation is established under EU pharmaceutical law:
- Article 57(2) of Regulation (EC) No 726/2004
- Directive 2001/83/EC, as amended by Directive 2010/84/EU (EU pharmacovigilance legislation)
The EMA relies on this data to assist with regulatory decision-making, data analysis, and public communication efforts.
What is the purpose of medicine data submission to the EMA?
The primary goal of submitting medicine-related data is to create a comprehensive database of all medicines approved for use within the European Union (EU) and European Economic Area (EEA). This includes both centrally authorised medicines through the EMA and those approved at the national level.
The European Medicines Agency (EMA) utilises this data for multiple purposes, such as:
- Data Analysis and Signal Detection
- Regulatory and Legal Compliance
- Improving Stakeholder Communication

What obligations does each MAH have regarding medicine data submission?
The submission of medicine-related data by marketing authorisation holders is a mandatory requirement introduced under the pharmacovigilance legislation (Directive 2010/84/EU and Regulation (EU) No 1235/2010, Article 57(2) of Regulation (EC) No 726/2004, Good Pharmacovigilance Practices (GVP) Module VI).
Any new marketing authorisation granted after 2 July 2012 must be reported to the EMA within 15 calendar days of being notified by the national authority.
Changes to a marketing authorisation—such as variations, transfers, renewals, suspensions, revocations, or withdrawals—must be communicated to the EMA no later than 30 calendar days after the changes are approved.
Moreover, the European Medicines Agency (EMA) is migrating medicinal product data from xEVMPD to the Product Management Service (PMS) under ISO IDMP standards. Improving data quality and interoperability. The PMS offers improved data management capabilities, including a user interface and API access, facilitating better regulatory processes and data reuse across the medicinal product lifecycle. This change is crucial for regulatory compliance and requires MAHs to standardise their data.
What is Art. 57 / xEVMPD service?
At SciencePharma, we provide comprehensive support in reporting and updating medicinal product data in the xEVMPD system – for both authorised products already on the market and newly registered ones. We offer full assistance in preparing, verifying, and submitting data, ensuring compliance, timeliness, and regulatory peace of mind. Our services include, among others:
- Preparation and registration of products in the xEVMPD database,
- Verification of data compliance with EMA and IDMP requirements,
- Updating information and submitting changes within the required timelines (e.g., following MA variations),
- Consulting on data migration to the PMS (Product Management Service) system,
- Data quality audits and development of corrective recommendations.
Who is this service for?
Our offer is directed at pharmaceutical companies (MAHs) that hold marketing authorisations in the EU and EEA countries, regardless of whether the product was approved through national, MRP/DCP, or centralised procedures.
What does the client gain from this service?
- Assurance of compliance with EMA requirements and applicable EU regulations – avoiding the risk of penalties and delays,
- Timely reporting of changes – in line with legal requirements, without burdening internal teams,
- Professional technical and regulatory support – simplifying complex processes,
- Reduction of data errors – through our audits and validation procedures,
- Readiness for future changes, such as migration to the PMS (Product Management Service) system,
- Relief for client teams and time savings – we handle the entire process.
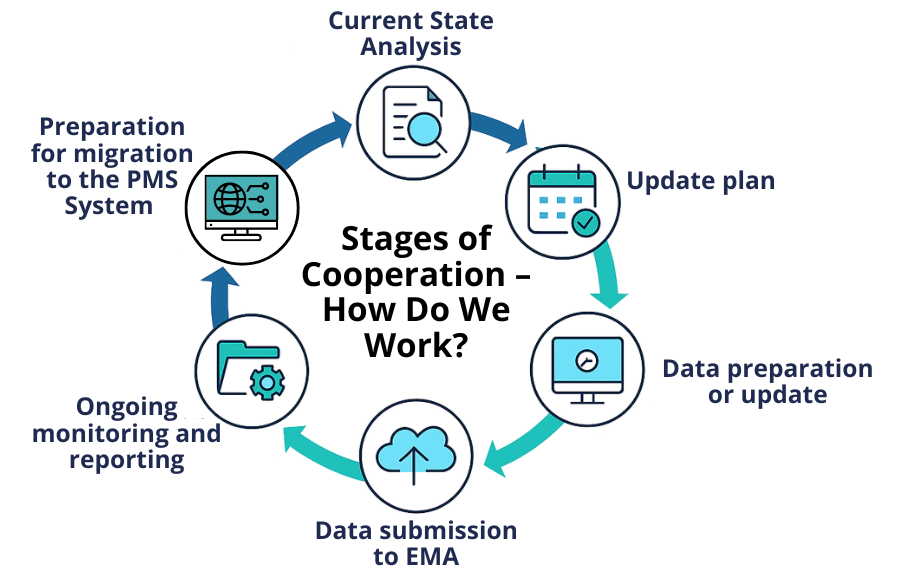
Stages of Cooperation – How Do We Work?
- Current State Analysis – verification of data completeness in xEVMPD
- Update Plan – action schedule and required data completion
- Data Preparation or Update – in line with applicable requirements
- Data Submission to EMA – reporting in the required format and confirmation of correctness
- Ongoing Monitoring and Reporting – tracking changes throughout the product lifecycle
- Preparation for Migration to the PMS System – data standardisation and consulting
Why Choose Us?
With years of experience and a team of dedicated experts, we provide comprehensive support throughout the entire product lifecycle. We manage everything from insertions and updates to handling variations, transfers, renewals, suspensions, revocations, and withdrawals with precision and care. Our services go beyond operational execution – we also offer strategic consulting, training, and compliance audits in this area.

What sets us apart?
Proven Experience – Our long-standing presence in the market speaks for itself. We have completed hundreds of operations in the xEVMPD database for both centrally and nationally authorised products. Our extensive experience covers a wide range of projects. We work with companies of all sizes – from local entities to global market leaders.
Expert Team – Our specialists have many years of experience working with the xEVMPD database and possess in-depth knowledge of EMA regulations, including ISO IDMP standards and GVP guidelines. They regularly participate in training sessions and stay up to date with regulatory changes, ensuring the highest level of expert support.
Up-to-Date Processes – We continuously adapt our workflows to meet the latest industry standards and regulatory requirements. We operate according to updated procedures aligned with EMA expectations and ensure the full auditability of all activities.
Complete Lifecycle Coverage – We offer comprehensive support across all processes related to the product lifecycle. We manage the entire data handling process in xEVMPD – from the submission of new products, through updates following marketing authorisation changes, to support during data migration to the PMS system. We take a proactive approach by monitoring changes in marketing authorisation statuses and reminding clients of upcoming regulatory obligations.
Partner with us and rest assured that your product is in expert hands, every step of the way.
Why us?
SciencePharma delivers professional pharmacovigilance support throughout the entire life cycle of a medicinal product. Our services are meticulously tailored to meet each client’s requirements, ensuring flexibility and precision. Through a personalised approach and the implementation of an advanced drug safety management system, we enable our clients to focus on the strategic development of their products while maintaining the highest standards of patient safety and regulatory compliance.
Contact us to find out how we can support your organisation in ensuring full regulatory compliance and the safety of pharmacotherapy.
References:
- https://www.ema.europa.eu/en/documents/other/data-submission-authorised-medicines-european-union-outlines-article-572-regulation-ec-no-7262004_en.pdf
- https://www.ema.europa.eu/en/data-medicines-iso-idmp-standards-post-authorisation/extended-eudravigilance-medicinal-product-dictionary-xevmpd-training
- https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/data-medicines-iso-idmp-standards-post-authorisation/data-submission-authorised-medicines-article-57