
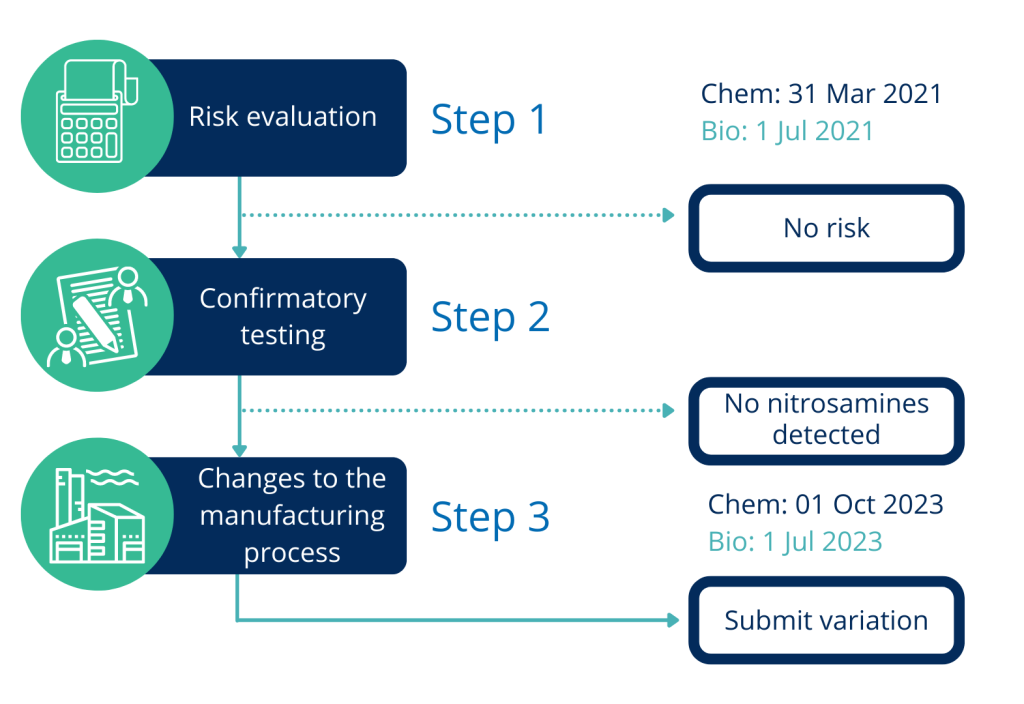
In June 2018 it was recognized that some active substances used in manufacturing of medicinal product contained undesired levels of potentially carcinogenic nitrosamine impurities (e.g. NDMA, NDEA). Immediately afterwards, EU regulators started to establish ways of minimizing that risk. As a result, MAHs were requested to review their manufacturing process and evaluate the risk of the presence and possible sources and formation of nitrosamine impurities in their medicinal products (CHMP opinion under Article 5 (3) of Regulation (EC) No. 726/2004). From that point onwards, MAH should subsequently inform the competent authorities about the outcome of such risk assessment. Depending on the outcome, if the presence of impurity is indeed of a concern, further steps, such as confirmatory testing or even changes in the manufacturing process and subsequent post-approval variations, may be required.
Is your deadline due? Chemical, biological or new applications.
Initially, the request to perform risk analysis on the presence of nitrosamines was forwarded to MAHs, whose already registered products contain chemically synthesized API. The deadline for submission of risk assessment outcome for these products has already passed on 31 March 2021. However, if concerned, MAHs have not yet performed such analysis, they should prioritize such activity as soon as possible.
Later on, in July 2020, the EMA request was expanded to also include products containing biological API, for which the deadline is very close – 1 July 2021. Regardless of the deadlines for existing MAs, it is worth to remember that risk assessment for the presence and possible sources and formation of nitrosamine impurities is required for all new applications. It is important to highlight this fact, to raise awareness of MAHs, holders of MIAs, CEPs and ASMFs.
Risk evaluation should also be performed for radiopharmaceuticals, but as it stands for now, does not need to be done for herbal medicinal products.
What are nitrosamines at all?
Nitrosamines (N-nitrosamines) are organic chemical compounds consisting of a nitroso group (NO+) bonded to a deprotonated, usually alkyl-group based amine. However, in general, nitrosamines refer to any molecule containing the nitroso functional group. Although they are present in some foods, drinking water supplies, or air, their presence in medicines is still considered unacceptable, since most of nitrosamines are considered carcinogenic (1).
The most popular nitrosamines – N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) belong to a group of highly potent mutagenic carcinogens, that have been classified by World Health Organization International Agency for Research on Cancer (WHO IARC) as probable human carcinogens (2,3).

Figure 2. General structure of nitrosaminesWhat triggers nitrosamines formation?
Scientific community identified a number of root causes of nitrosamine formation and contamination of drug medicinal products. The manufacturing conditions, that may promote nitrosamine impurities formation during API synthesis, include acidic pH and presence of certain types of raw materials, starting materials, and intermediates (especially those that contain nitrogen (III)). It can be highlighted that the use of sodium nitrite (NaNO2), or other nitrites, in the presence of secondary or tertiary amines is a potential cause of nitrosamine formation. However, it is also important to notice that, a sheer presence of amines alone is not sufficient for the nitrosamines to form. This is why, although biological products contain many amines, the risk of nitrosamine formation is often rather negligible.
Among the tertiary amines there are compounds that have nitrosamine-forming properties (e.g., triethylamine, diisopropylethylamine (DIPEA)). It is also worth noting that tertiary amines are also common functional groups in many APIs and active ingredient precursors. Secondary amines can be present in reagents and solvents as impurities or degradants. They may also be part of reagents, solvents, APIs, their degradants, and precursor structures. For example, amide solvents can degrade to secondary amines which are known sources of nitrosamines (such as N,N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), or N,N-dimethylacetamide (DMA). It is important to notice that, in most of confirmed cases of nitrosamine contamination of APIs, the nitrite source and amine have been used in the same step. However, cases are known where, despite extensive purification operations, sodium nitrite is carried over from previous steps and reacts with an amine to generate a nitrosamine impurity.
As mentioned earlier raw materials, when contaminated, may be an important source of nitrosamines. Recycled solvents, reagents, and catalysts, may pose a risk for nitrosamine formation due to the presence of amines in the waste streams sent for recovery and the subsequent quenching of these materials with nitrous acid to destroy residual azide, without adequate control of nitrosamine formation or adequate purification. Examples of recycled materials observed to be contaminated with nitrosamines include orthoxylene and tributyltin chloride (used as a source of tributyltin azide). It has also been suggested that N,N-dimethylformamide (DMF) could be contaminated in this way.
Nitrosamines can also be introduced to the medicinal products via contaminated starting materials, including intermediates, supplied by vendors that use processing methods or raw materials causing formation of nitrosamines. For example, nitrites are known impurities in raw materials, including reagents, solvents, and excipients used in finished products.
The nitrosamines precursors include:
- primary, secondary and tertiary amines (aliphatic or aromatic);
- 4th order ammonium salts;
- pesticides;
- drugs with amino groups (eg. penicillin, ephedrine, promethazine);
- compounds containing groups with a nitrogen atom (III).
The kinetics of the nitrosamine formation reaction is influenced by:
- concentration of substrates (especially the presence of “nitro” species);
- basicity of the amine (the higher the pKb, the more basic the amine);
- pH of the environment (acidic pH, typical pH 3.3);
- temperature (in general, its increase increases the concentration of the product);
- presence of reaction catalysts, eg. formaldehyde, nitrogen oxides or thiocyanates

Brief guide through risk assessment
The review of the manufacturing conditions for API(s) and the final medicinal product should generally be based on the risk analysis performed by the manufacturers of API(s) and product, as well as on the current chemical/biological knowledge about nitrosamines.
- Highlighting the stages, where amines or nitrite agents are used or demonstrating its lack.
- Analyzing raw materials used, like quaternary ammonium salts.
- Taking into account and reviewing any catalysts used.
- Demonstrating presence or lack of nitrogen-based impurities.
- Considering purification methods in relation to their the potential (or real) effectiveness in nitrosamine impurity removal (compare with [4]).
Last, but not least, the storage and transport conditions and packaging materials should be also considered, as they too may be a potential hazard source.
Whom to share the news?
Once the risk analysis is performed, its outcome should be sent to respective Agency (for national authorities) or Agencies (RMS, CMS for DCP/MRP procedures) or EMA (centrally registered products). If there is no risk, no further steps are required. The first step requires only the outcome of the risk assessment while the full risk analysis may be requested by the Agency. Relevant templates are provided on EMA website. If a risk is detected then, the following steps should be employed such as confirmatory testing or even change in manufacturing process and subsequent variation.
Summary
Nitrosamines are chemical substances that are considered potentially carcinogenic. Although they are occurring in the environment (food, water, air) their presence in medicinal products is considered unwanted. EU Agencies have established multi-step plan for MAH to confirm the lack or reduce the risk of nitrosamine formation. It starts with the obligatory risk assessment for the presence and possible sources and formation of nitrosamine impurities in medicinal products that contain chemically synthesized and biological API, and subsequently, informing Agencies of the outcome. The deadline for the chemical products has passed (31 March 2021) and for the biological is approaching (1 July 2021). The burden of such risk analysis is laid on MAH. Each MAH should review all of its registered products (even if not commercialized). At the same time, all new applications should be supplemented with the outcome of such risk assessment. Risk assessment should be available on request, but does not need to be added into a dossier. It should be made case-by-case and be based on review of a process and scientific knowledge. It should cover the manufacturing conditions that may promote nitrosamine impurities formation during API synthesis with special consideration of the pH and presence of certain types of raw materials, starting materials, and intermediates (especially those that contain nitrogen (III)).
*article was updated on 03.08.2022.
*Please note that provided deadlines are subject to EMA updates.