
Registration of medicinal products is a key process that ensures that all medicines distributed and sold in the European Union (EU) meet established quality, safety and efficacy standards. Therefore, regulatory systems across the EU have been carefully designed to thoroughly evaluate medicinal products before granting marketing authorisation (MA).
Applicants can use several ways to obtain EU MA:
- the national procedure,
- the decentralised procedure,
- the mutual recognition procedure,
- and the centralised procedure (CP).
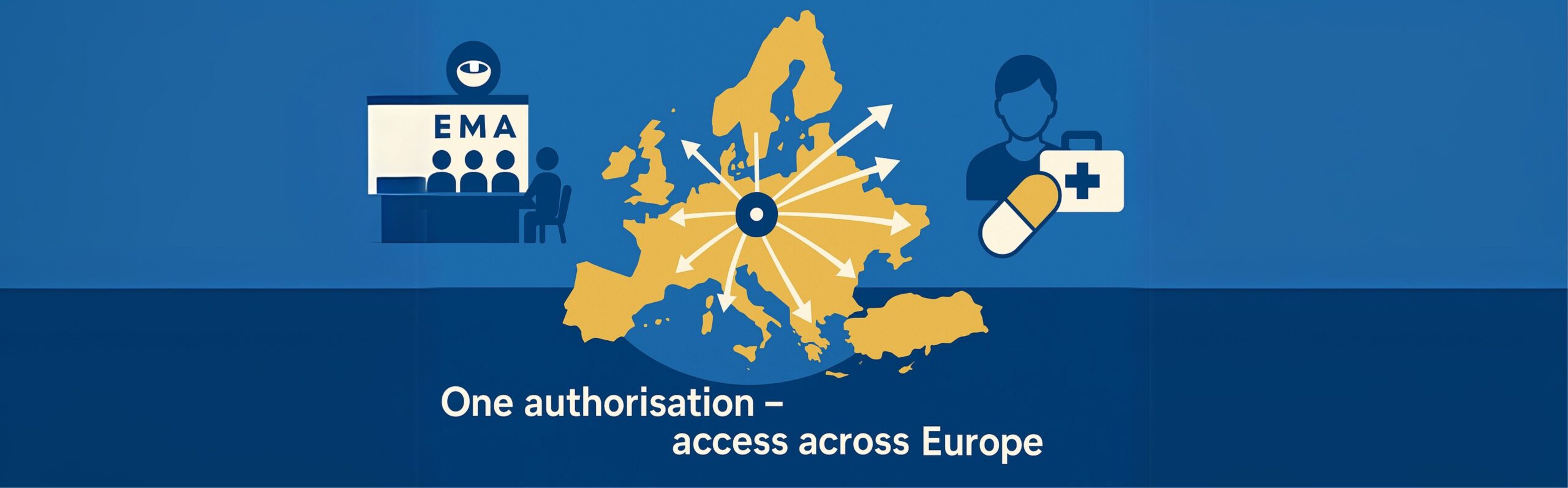
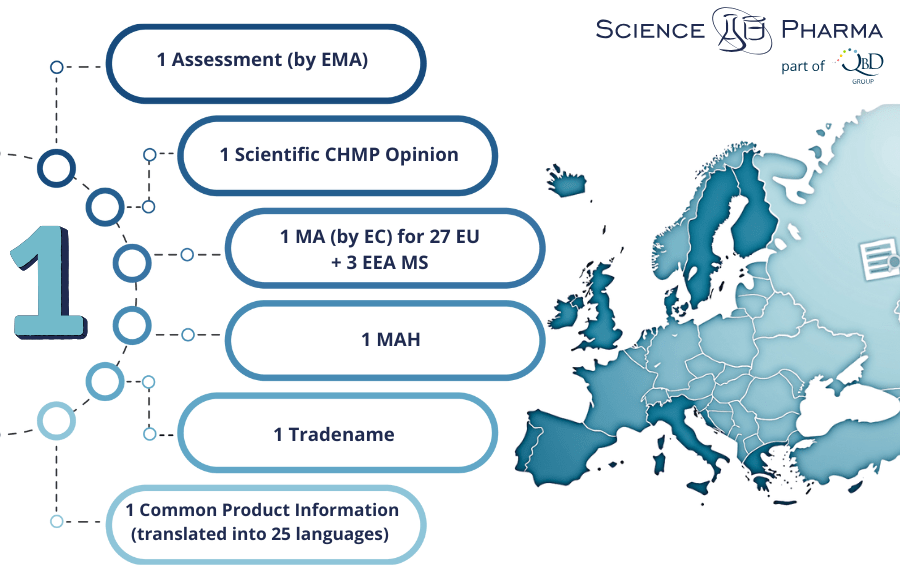
Among them, the CP allows the marketing authorisation holder (MAH) to market the medicine and make it available to patients and healthcare professionals throughout the EU and the three European Economic Area (EEA) Member States (MS): Iceland, Liechtenstein, and Norway, based on a single MA.
The core of the regulatory system is the European Medicines Agency (EMA), based in Amsterdam. The EMA coordinates the evaluation and monitoring of medicines in the EU and works with national competent authorities and specialised committees, such as the Committee for Medicinal Products for Human Use (CHMP), which is responsible for issuing scientific opinions on marketing authorisation applications.
The European Commission (EC) makes the final legal decision to grant or deny a centralised marketing authorisation based on the CHMP’s scientific evaluation. This two-step approach—a scientific evaluation by experts and a legal decision by the Commission—ensures both technical excellence and political accountability.
In the context of global pharmaceutical development, the CP provides companies with streamlined access to a market of approximately 450 million people, facilitating faster patient access to innovative therapies.
This article comprehensively discusses the centralised procedure, offering insights into the regulatory framework, advantages, challenges and practical strategies for successful authorisation.
Historical evolution of the CP
The CP and the EMA were introduced in 1995 under Council Regulation (EEC) No 2309/93 of 22 July 1993, laying down Community procedures for the authorisation and supervision of medicinal products. In October 1995, the first medicine authorised under the CP was the fertility treatment Gonal-F.
Initially, the centralised procedure was limited to biotechnology-derived products and certain high-technology medicines. Over time, its scope expanded significantly:
- 2000: Orphan medicinal products for rare diseases were incorporated under the centralised route via Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products;
- 2007: Advanced therapy medicinal products were brought into scope through Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products;
- 2012: Establishment of the Pharmacovigilance Risk Assessment Committee to strengthen post-marketing surveillance.
The CP’s success in promoting consistency, efficiency, and public health protection has led to its current pivotal role within the EU pharmaceutical regulatory system. It has been particularly instrumental in facilitating access to therapies for rare diseases, personalised medicines, and pandemic responses.
Today, the great majority of new, innovative medicines pass through the centralised authorisation procedure to be marketed in the EU. Applicants seeking to use the centralised procedure voluntarily must request eligibility confirmation from the EMA before submitting their marketing authorisation application.
The legal framework governing medicinal products for human use in the EU
The EU has established a harmonised regulatory framework for medicinal products to ensure that all member states follow consistent standards. The basis for this regulatory framework is several key pieces of legislation:
- Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency
Governs the CP and establishes the EMA.
- Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use
Creates a Community code relating to medicinal products for human use, including requirements for marketing authorisation, manufacturing, distribution and pharmacovigilance.
- Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use
Regulates clinical trials, ensuring that research conducted in the EU meets harmonized standards of ethics and scientific integrity.
The CP is complemented by procedural guidelines issued by the EMA and European Commission (EC), covering detailed operational aspects such as validation, scientific advice, accelerated assessment, and pharmacovigilance obligations.
Recent regulatory initiatives, such as the “Pharmaceutical Strategy for Europe” and updates to pharmacovigilance legislation, demonstrate the EU’s ongoing commitment to adapting its system to scientific progress, emerging health risks and societal needs.
In November 2020, the EC launched the Pharmaceutical Strategy for Europe, aiming to modernise the regulatory framework and address persistent challenges.
Key Objectives:
- Promote innovation: Foster the development of novel therapies, including advanced and personalised medicines.
- Strengthen crisis preparedness: Build resilience against shortages and supply chain disruptions.
- Improve access and affordability: Ensure equitable access to high-quality medicines across all Member States.
- Promote environmental sustainability: Address the environmental impact of pharmaceutical production and disposal.
Expected Changes Affecting the CP:
- Streamlining the authorisation process for high-need medicines.
- Enhancing the role of real-world data and digital health technologies.
- Revising incentives for orphan and paediatric medicines to balance innovation with affordability.
- Promoting regulatory agility through adaptive pathways and rolling submissions beyond emergency contexts.
The Pharmaceutical Strategy recognises the centralised procedure as a key mechanism for driving innovation, safeguarding public health, and delivering timely access to critical therapies across Europe.
Step-by-step: eligibility and pre-submission activities
Before initiating a marketing authorisation application (MAA) under the CP, companies must ensure that their product qualifies for this regulatory pathway. The EMA provides a formal eligibility confirmation process to determine whether the product falls within the mandatory or optional scope outlined in Regulation (EC) No 726/2004 and below:
The scope of the CP includes:

Mandatory medicinal products:
- Medicinal products developed by biotechnological processes.
- Orphan medicinal products are designated for rare diseases.
- Advanced therapy medicinal products (ATMPs) such as gene therapies, somatic cell therapies, and tissue-engineered products.
- Products containing new active substances for the treatment of serious diseases, including HIV/AIDS, cancer, neurodegenerative disorders, diabetes, and autoimmune diseases.
Optional medicinal products:
- Products representing significant therapeutic, scientific, or technical innovation.
- Products whose authorisation under the CP would be in the interest of public health at the Union level.
Eligibility Request:
- Applicants must submit a written request to EMA at least six to seven months before the planned MAA submission date.
- The request must include a detailed justification supported by scientific data, demonstrating how the product meets the scope criteria.
- EMA’s Scientific Advice Working Party (SAWP) or dedicated assessment teams review the request and issue an opinion.
Key Eligibility Scenarios:
- Mandatory Eligibility: For products involving biotechnology, advanced therapies, orphan diseases, or specific major public health threats.
- Optional Eligibility: If a product represents a significant therapeutic, scientific, or technical innovation or if centralised authorisation serves the Union’s public health interests.
Pre-Submission Meetings:
- Applicants are strongly encouraged to request a Pre-Submission Meeting with EMA staff, which typically occurs 7–9 months before planned submission.
- Discussions focus on dossier structure, procedural logistics, regulatory strategy, and specific scientific concerns.
Other Important Pre-Submission Activities:
- Scientific Advice: Companies may seek early scientific advice to align their clinical and non-clinical development with regulatory expectations.
- Orphan Designation: If the product targets a rare disease, an application for orphan designation must be submitted early to benefit from incentives such as reduced fees and market exclusivity.
- Pediatric Investigation Plan (PIP): Compliance with EU pediatric regulations is mandatory unless waivers are granted.
Engaging early with EMA significantly reduces the risk of procedural delays and helps applicants refine their regulatory strategies to align with both EMA and broader EU expectations.
Step-by-step: dossier preparation and evaluation timeline
A successful centralised application hinges on the quality, completeness, and organisation of the MA dossier. The dossier must follow the Common Technical Document (CTD) format, structured into five modules:
- Module 1: Administrative and regional information specific to the EU (e.g., application forms, product information, legal documentation);
- Module 2: Summaries and overviews of the technical modules (quality, non-clinical, clinical);
- Module 3: Quality documentation;
- Module 4: Non-clinical study reports;
- Module 5: Clinical study reports.
Once the MAA is validated, the scientific evaluation process begins, coordinated by the CHMP at the EMA.
Key Features of the Scientific Evaluation:
- Appointment of Rapporteurs: Two rapporteurs (primary and co-rapporteur) from different national competent authorities are appointed to lead the assessment.
- Scientific Advisory Groups (SAGs): Experts may be consulted for specific therapeutic areas (e.g., oncology, infectious diseases).
Evaluation Timeline:
- Day 0: Start of scientific assessment.
- Day 80: Rapporteurs submit their Assessment Reports; the CHMP issues a List of Questions (LoQ) to the applicant.
- Day 120: Interim discussion; decision on whether the application can proceed or needs major revisions.
- Clock Stop: Applicant responds to LoQ. Time is flexible, typically 3–6 months.
- Day 121: Restart the assessment after the applicant’s responses.
- Day 180: Second Assessment Reports and List of Outstanding Issues (if any) are issued.
- Day 210: Final CHMP opinion (positive or negative) is adopted.
Additional Steps:
- Oral Explanations: In complex cases, applicants may be invited to an oral hearing before the CHMP to address concerns;
- Referral Procedures: If major concerns remain unresolved, a referral to the CHMP under Article 29 may be initiated.
A positive CHMP opinion leads to a binding decision by the EC within 67 days. A negative opinion results in refusal of MA, though applicants may request a re-examination.
Granting a marketing authorisation is only the beginning. Lifecycle management ensures that the product continues to meet safety, quality, and efficacy standards throughout its commercial existence.
To sum up
The CP remains an indispensable mechanism for ensuring that innovative, high-quality medicinal products swiftly and equitably reach patients across the EU. Its strengths lie in scientific rigour, regulatory transparency, and harmonisation of standards across diverse Member States.
However, successful navigation of the CP demands meticulous preparation, proactive regulatory engagement, and a comprehensive understanding of evolving scientific and policy landscapes.
Looking forward, the centralised procedure is set to evolve further in response to technological innovations, global public health challenges, and new policy priorities under the EU Pharmaceutical Strategy.
For companies committed to bringing transformative therapies to European patients, mastering the CP will remain not just a regulatory requirement but a critical strategic advantage.
References:
- European Comission, https://health.ec.europa.eu/medicinal-products/legal-framework-governing-medicinal-products-human-use-eu/authorisation-procedures-centralised-procedure_en;
- European Commission Pharmaceutical Strategy for Europe (2020);
- EMA Procedural Guidance Documents;
- European Medicines Agency, https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines#scope-of-the-centralised-procedure-10943;
- European Medicines Agency, https://www.ema.europa.eu/system/files/documents/presentation/wc500201043_en.pdf;
- Shaik Azeem, Syed Kashif, Md Meezan, Nizamullah G N, S.B Puranik, EMA, EDQM and CEP, Journal For Innovative Development in Pharmaceutical and Technical Science (JIDPTS), Volume: 4, Issue: 6, June: 2021.