

Module 3 is a part of the Common Technical Document (CTD) that contains all the quality data needed to the registration of a human medicinal product. The CTD is a special documentation format used for the submission of registration information of medicinal products to regulatory authorities. In 2003, it became required for all medicinal product submissions in the EU, Japan and recommended in US. It is worth noting that more and more other countries also adjust to this format, for example Switzerland, Turkey, Brazil, Singapore, Canada, Republic of Korea, China or Chinese Taipei.
The CTD format applies to all categories of medicinal products and also for all types of applications planned to be submitted to regulatory authorities. It consists of 5 modules: Modules 2 to 4 are the formal part of CTD and are common for all regions, while module 1 is regionally dependent and is not recognized as a part of CTD. Schematic diagram of the organization of the CTD is presented in the Picture 1.

Module 1 contains regional information, module 3 describes quality of active substances and medicinal products, module 4 and 5 concern non-clinical and clinical studies, respectively. Module 2 contains overviews and summaries of modules 3, 4 and 5. The CTD format is intended to improve organization of documents and therefore allows harmonization of data across different products. However it does not itself indicate what exact studies and data should be submitted in each module for successful validation and approval. The detailed content requirements of each section may depend on regional requirements. In this article we focus on module 3 – that is quality and the general type of data that it should contain!
Picture 1. Schematic diagram of CTD triangle.

Module 3 in CTD format
Module 3 as the part of CTD is divided into sections – section 3.1 with table of contents of module 3 and section 3.2 with body of data. The latter is further grouped into section 3.2.S divided into 7 subsections that all refer to quality of active substance(s) and section 3.2.P divided into 8 subsections that all refer to quality of a medicinal product. The CTD format precisely describes the order in which each qualitative data should be presented and allows for a transparent and legible presentation of all the required quality data. Moreover, module 3 contains section 3.2.A with appendices, 3.2.R with regional information and 3.3 that includes literature references. The scope of sections of module 3 is visualized in the Picture 2.
Picture 2: The scope of sections of module 3
What is the quality of the medicinal product in registration dossier?
Module 3 is one of the fundamental parts of the registration dossier. It contains all the essential information about the quality of the medicinal product and its active substances. The appropriate quality of a medicinal product guarantees patient safety, which is why it should be properly controlled from the beginning of development of medicinal product through its manufacturing and during shelf-life. Enormous amount of quality data is required from the Applicant to obtain a marketing authorization. Exact structure of section 3.2.S and 3.2.P with highlighted subsections is presented here.
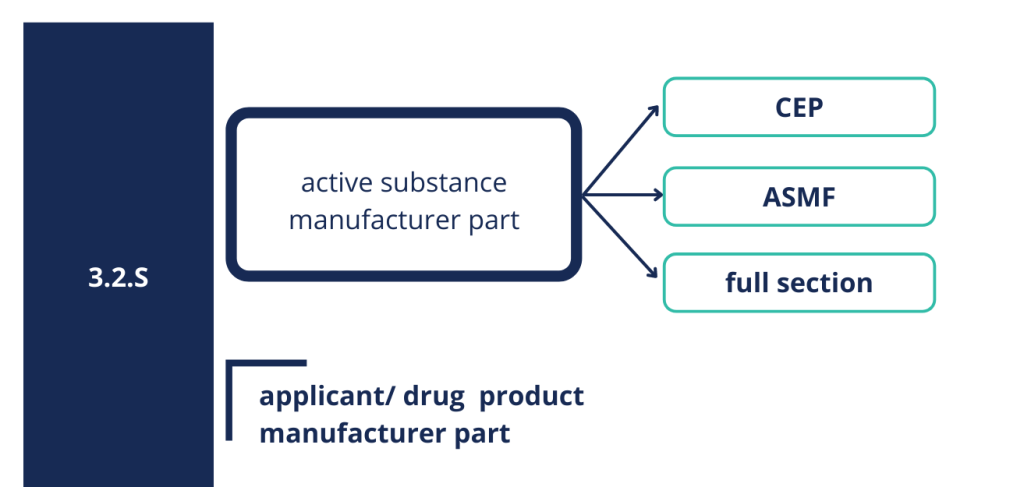
Section 3.2.S related to the quality of the active substance can be presented as full section eighter for biological or biotechnological medicinal products, however most often it is presented in the form of active substance master file (ASMF) or certificate of suitability (CEP) issued by European Directorate for the Quality of Medicines (EDQM). CEP proves that the quality of API is in compliance with the requirements described in the relevant monograph of the European Pharmacopoeia (Ph. Eur.). ASMF consists of two parts – open part accessible by the Applicant, and restricted part not available to the Applicant. The latter contains confidential information and therefore is directly submitted by the active substance manufacturer to the regulatory authority.
Picture 3: Structure of 3.2.S section.
It should be remembered that CEP and ASMF cover part of the quality data but the Applicant takes full responsibility for the quality of the active substance, and is therefore also obligated to control appropriate quality of the active substance. The Applicant has to ensure that he has access to all relevant information concerning the manufacture of the active substance, in order to ensure that is capable to take responsibility for the quality of the active substance in the final medicinal product. In principle, the specification of active substance provided by the Applicant/drug product manufacturer should be identical to that of the ASMF or the CEP holder and if there are any differences, appropriate justification should be provided. In general, the set of information and experimental data presented in section 3.2.S proves and confirms the appropriate quality of the active substance that will be used in the production of the medicinal product.
Section 3.2.P contains all chemical and pharmaceutical information needed to prove the highest quality of the medicinal product. In this short article we would like to show how many data needs to be presented in section 3.2.P to be assessed by the regulatory agencies and how huge cooperation and commitment of different teams is required to ensure appropriate quality of a medicinal product. The scope of studies to be performed isspecified in the relevant guidelines. Section 3.2.P includes qualitative and quantitative information on the composition of the medicinal product, along with excipients and describes appropriate selection of the composition of the medicinal product as well as the compatibility of individual ingredients. Pharmaceutical development, that makes up a large part of section 3.2.P, contains information on all the studies conducted to choose appropriate formulation (e.g. tablets), to choose appropriate manufacturing process (e.g. dry-tableting) with critical process parameters (e.g. hardness) and to choose container closure system (e.g. blister) which allows to maintain an appropriate shelf life of medicinal product and protects against the influence of the external environment. What tests will be presented in quality part of dossier depends on many factors including dosage form (solid, liquid, oral, parenteral, topical, inhalation etc.), registration category (generic, hybrid, WEU etc.) or target population (children, adults, older people). In case of the multi-dose product, preservatives deserve a special attention and their use should be properly justified in dossier (in particular for products intended to use in child population). It has to be proven that the minimum concentration of antimicrobial preservatives which gives the required level of efficacy has been used. Depending on the category in which the product is registered, this section may need to contain appropriate comparative tests with reference product. Important data relating to the quality of medicinal product is information on impurities coming from both the synthesis of active substance and manufacturing process. Impurities should be properly identified and then, if needed, controlled to keep them at an appropriate level that does not compromise patient safety. When it comes to impurities, the special attention deserves the need of performing the risk analysis for the presence and possible sources of nitrosamine impurities in medicinal products.The details are covered in our last article “Are nitrosamines still a concern for a drug product?” – we encourage you to read it! All these data have to be analyzed and described in respective sections.

The quality of the product is also a result of appropriate manufacturing process which must be precisely described at defined scale. The critical steps, which are those stages of manufacturing that may have a critical impact on the quality of the final product, have to be identified and appropriate in-process control should be implemented. The entire manufacturing process must be validated. It is worth noting that each manufacturer including laboratories responsible for batch testing in EU as well as batch certification site(s) should be provided in registration dossier. The medicinal product should be tested in accordance with the provided specification and in accordance with the analytical methods described in the section. Methods should be appropriately selected and, if needed, especially for non-pharmacopoeial methods, validated. Appropriate selection of container closure ensures stability of the product over the appropriate shelf life, which means that the product maintains its characteristic and activity as long as the patient administers it within the expiration date. If a medicinal product is to be administrated with help of a dosage device, such as syringe, cup, inhaler, etc. additional studies may be required, depending on the type of device, such as droplet size distribution or uniformity of dispensed dose for a particular dosage form (e.g. syrup, suspension, solution, powder, spray). In addition, in the case of multi-dose containers, it is required to prove that medicinal product has appropriate in-use stability (the time a medicine can be used after first opening). The information presented above is only a fraction of what the Applicant should take into account before submitting registration dossier to the regulatory authority in order to obtain a marketing authorization for its medicinal product. The key to success lies in looking at the developed product very broadly, from different angles, keeping the regulatory aspects in mind.

Where to find relevant information?
Prior to start of the development work, followed by respective studies and subsequent registration of human medicinal product, the Applicant should define the target market(s) where the medicinal product will be registered. Depending on the region, a different range of quality data may be required in module 3. In European procedures, it is the European Medicines Agency (EMA) that is responsible for registration process. The Agency provides guidance and support to medicine development and dossier submission. In the US it is the Food and Drug Administration (FDA) that serves similar role. If a medicinal product is registered under national procedures, national medicinal product regulations will apply. Questions “how detailed the data should be presented and whether all available data must be added to the dossier” are frequent and highly important and by such should always be answered when preparing the module 3.
Too detailed descriptions may pose a challenge during a life-cycle of a product, i.e. any future post-registration variations that may affect the dossier should be considered as any change can be associated with additional time and costs.
Please note, that as a rule, Good Manufacturing Practice (GMP) issues are not strictly considered in the registration dossier and so were not covered by this article. However, it should be remembered that GMP is a practice that must be followed during manufacturing and control of any active substance and medicinal product. The aim is to operate at the highest possible quality standards. All the guidelines and medicinal product regulations should be read very carefully and in conjunction with each other. In case something is unclear, it is worth consulting a professional company and/or applying for an official scientific advice to appropriate Agency.
Let’s go deeper into Module 3
Let’s imagine for instance a product in a form of coated tablets consisting of two APIs – what information should be taken into account while preparing a respective Module 3? Suppose active substance manufacturers have valid CEPs – these certificates will be presented to prove appropriate quality of both APIs. Moreover, a drug product manufacturer also controls appropriate quality of both active substances and will describe it in section 3.2.S.4.
When we think about section 3.2.P and final medicinal product, we need to know and accurately describe composition of medicinal product, including excipients and its role (e.g. binder, filler, lubricant) in section 3.2.P.1. The ingredients of the product must be compatible with each other which should be confirmed by appropriate developmental tests (3.2.P.2) and stability studies (3.2.P.2, 3.2.P.8). Moreover, we should know the impurities’ profiles (3.2.P.2) to establish appropriate specification. The final product specification (3.2.P.5.1) should take into account pharmacopoeial requirements for dosage form: tablets and other parameters mentioned in appropriate guidelines, i.e. for solid oral dosage forms.

The analytical methods, for example chromatographic ones, that are used to test the tablet – product should be described (3.2.P.5.2) and validated for specificity, accuracy, precision, etc. (3.2.P.5.4). It is a similar case for the entire manufacturing process which should be described (3.2.P.3.3) and validated (3.2.P.3.5). The manufacturer needs to check whether any of the process parameters is a critical process parameter (CPP), which may affect products critical quality attributes (CQA). If so, they should be defined and appropriate in-process controls should be introduced (3.2.P.3.3). For example, let’s imagine that a parameter speed of mixing subsequently and critically affects uniformity of tablets. It makes the parameter critical and therefore it should be maintained within appropriate range. For tablets we should also present dissolution profiles, which are an in-vitro gate to the in-vivo world. These studies should be provided using appropriate apparatus and mobile phases, especially considering physiological pH conditions. The selection of the primary and secondary packaging is also crucial – if the described tablets are packed in blisters, we have to prove that such container closure system allows the product to be stable throughout its shelf-life, if they are packed in multi-dose containers, like HDPE bottles, we have to present in-use stability study.
Whenever Module 3 is prepared, we should approach each product individually! As in the above case of coated tablets, the release profiles are an important aspect, but in the case of syrups such tests would be unnecessary and even not possible. There are some general rules, as to what type of information should be covered in module 3. However, there are no detailed rules for each type of product and so each one should be approached with due care.
To the end
Determining the scope of quality data that should be included in the module 3 of registration dossier it is both difficult and time consuming process which requires extensive knowledge and experience. The multitude of medicinal product regulations may become challenging to follow. It often crosses various scientific and regulatory areas and therefore various teams should closely cooperate when working on defining the scope of quality data. Collecting the appropriate data and organizing it in a well-structured format is the end-goal in a process of assuring the highest quality and safety of a medicinal product.
The result of properly prepared registration dossier is a marketing authorization. We – patients – can be therefore quite confident, that the products we use are safe, effective and of sufficient quality.
Please also see our “CTD Module 3 – Quality service” to know the areas where we can help if you struggle with building your own module 3 or want to assess someone’s module 3, and many more.