

-
Rozwój produktu leczniczego
-
Badania kliniczne
-
Jakość
-
Audyty GxP
-
Usługi Rejestracyjne
-
Wsparcie przedrejestracyjne
- Regulatory roadmap
- Usługa Scientific Advice
-
Druki informacyjne
-
Dokumentacja rejestracyjna
- Audyt dossier
- Proces rejestracji
- Obsługa porejestracyjna
- Regulatory outsourcing
-
Wsparcie przedrejestracyjne
-
Pharmacovigilance
-
Usługa QP / Import
Przygotowanie części klinicznej dokumentacji rejestracyjnej, przegląd kliniczny
Składając wniosek rejestracyjny, Wnioskodawca zobligowany jest do dołączenia do niego oceny produktu leczniczego. Na taką ocenę składa się między innymi ocena jakości, ocena niekliniczna i kliniczna.
Cel i zakres dokumentacji klinicznej
Przygotowanie klinicznej części dokumentacji rejestracyjnej polega przede wszystkim na zebraniu i krytycznej ocenie danych klinicznych dostępnych dla wnioskowanego produktu leczniczego. Danych, które bezpośrednio odnoszą się do wniosku rejestracyjnego, tj. wyselekcjonowanych indywidualnie dla danego produktu leczniczego odnośnie strategii rozwoju, wskazań czy dawkowania.
W tym celu wykonywany jest wyczerpujący i kompleksowy przegląd kliniczny. Przegląd podsumowany jest zwięzłą dyskusją i interpretacją dostępnych danych.
Właściwa ocena kliniczna produktu jest kluczem do sukcesu rejestracyjnego.
Punktem wyjścia jest oczywiście Informacja dla Wnioskodawców, ang. Notice to Applicants, NTA.
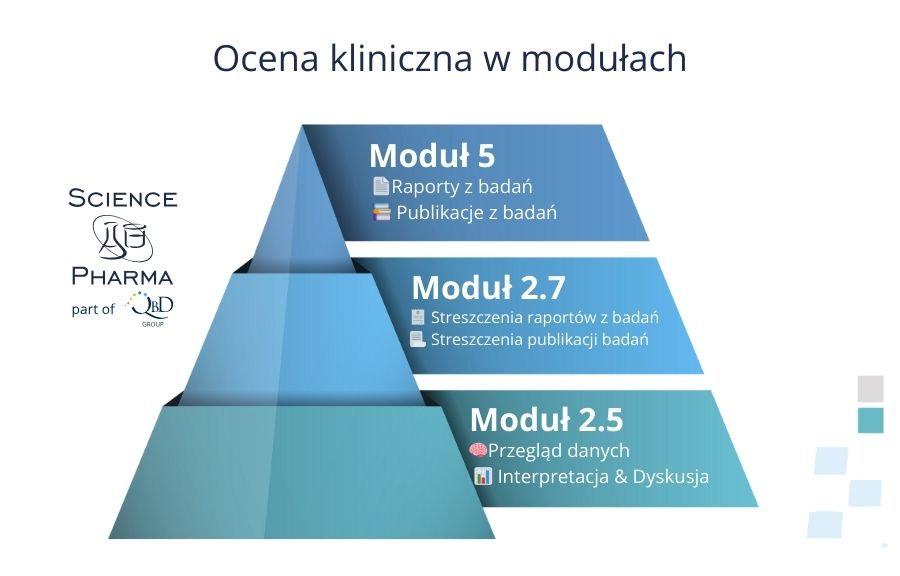
Ocena kliniczna zawarta jest w modułach: 5 (raporty z badań, publikacje badań), 2.7 (streszczenia raportów z badań, streszczenia publikacji badań) oraz 2.5 (przegląd danych, interpretacja i dyskusja).
Najistotniejsze elementy przeglądu klinicznego to:
- opis i wyjaśnienie ogólnego planu rozwoju klinicznego produktu leczniczego;
- opis i ocena klinicznych dowodów naukowych pod względem ich jakości (obejmujące m.in. metodykę badań, dobór pacjentów, czas trwania badań, wybór punktów końcowych, ocenę grupy kontrolnej, ocenę analizy statystycznej);
- ocena bilansu korzyści i ryzyka w oparciu o wnioski z badań klinicznych, w tym uzasadnienie proponowanego wskazania i dawkowania;
- analiza istotnych cech populacji pacjentów, w tym: stadium choroby, czynniki ryzyka wystąpienia choroby, grupy wiekowe, płeć;
- wskazanie sposobów ograniczenia potencjalnego ryzyka dla pacjenta wynikającego ze stosowania produktu;
- zaadresowanie ewentualnych problemów napotkanych w trakcie rozwoju produktu wraz ze sposobem ich zniwelowania lub wykazanie że zaistniałe problemy nie muszą stanowić przeszkody w trakcie rejestracji produktu leczniczego.
Moduły kliniczne w różnych ścieżkach rejestracyjnych
Podążanie za wskazówkami ujętymi w NTA nie jest jednak wystarczające. W różnych typach wniosków rejestracyjnych, moduły kliniczne będą prezentowały się nieco inaczej. Tutaj istotne jest uwzględnienie szeregu regulacji oraz wytycznych specyficznych dla danej strategii rejestracyjnej, a także dla substancji będących przedmiotem zainteresowania.
W typach wniosków opartych o badania własne, jak np. dla produktu oryginalnego przedstawia się program rozwoju klinicznego produktu leczniczego, w tym trwające i planowane badania kliniczne. Filarem tego typu wniosku są własne badania skuteczności i bezpieczeństwa rozwijanego produktu leczniczego. Uzyskane wyniki z badań własnych podlegają dyskusji w odniesieniu do danych literaturowych oraz wyznaczonych standardów leczenia opisanych w wytycznych klinicznych. Analizuje się podobieństwa i różnice w wynikach pomiędzy badaniami lub w różnych podgrupach pacjentów w ramach badań oraz ich wpływ na interpretację danych dotyczących skuteczności.
Do puli danych klinicznych własnych można dołączyć dowody naukowe pochodzące z baz informacji medycznej – istotnym jest jednak że stanowią one jedynie uzupełnienie i nie mogą to być główne badania dla produktu (mowa wówczas o tzw. mixed application).
Z kolei w typach wniosków gdzie referuje się do innego produktu leczniczego, jak np. generyk czy hybryda, kluczowe jest przedstawienie krytycznej analizy wszelkich istotnych kwestii związanych z biodostępnością produktu rozwijanego względem referencyjnego. W tych typach wniosków porównywalność biodostępności pomiędzy tymi produktami ma przełożenie na możliwość wnioskowania o skuteczności i/lub bezpieczeństwie produktu wprowadzanego do obrotu. Tutaj kluczowe jest syntetyczne ujęcie najważniejszych elementów badania biorównoważności lub udowodnienie że zostały spełnione warunki do zwolnienia z wykonywania takiego badania.
Przegląd kliniczny dla produktu o ugruntowanym zastosowaniu medycznym (w kategorii WEU, ang. well-established use) jest oparty zazwyczaj wyłącznie o dane literaturowe. Kluczowym jest jednak by dane te w pełni wspierały wybrane wskazanie i schemat dawkowania w określonej grupie docelowej, a także wykazanie, że mają one zastosowanie do wnioskowanego produktu. Dopuszczalne są pewne ekstrapolacje, jednak niezbędne jest w takiej sytuacji wieloletnie doświadczenie z Agencjami rejestracyjnymi by móc ocenić szanse na ich zaakceptowanie.
Wymogi dla produktów OTC – szczególna analiza bezpieczeństwa
Osobną kwestią jest planowana kategoria dostępności rozwijanego produktu. Jeśli przedmiotem zainteresowania jest dostępność bez recepty (OTC), oferujemy dokumentację kliniczną rozbudowaną o szczegółową ocenę bezpieczeństwa. Taka ocena ukierunkowana jest na wykazanie, że lek można bezpiecznie stosować bez nadzoru lekarskiego.

Dlaczego my?
Prowadzimy kompleksowe usługi rejestracji w Polsce i innych krajach europejskich. Bazujemy na wiedzy i umiejętnościach zespołu wykwalifikowanych ekspertów. O naszej skuteczności świadczą liczne sukcesy rejestracyjne, o których możesz przeczytać tutaj.
Korzyści współpracy
Zgodnie z życzeniem Wnioskodawcy dokumentację kliniczną możemy przygotować kompleksowo z innymi modułami dokumentacji, a także z drukami informacyjnymi dla produktu. Nie tylko przygotowujemy dokumentację – możliwe jest także zlecenie przeprowadzenie całego procesu rejestracyjnego. Wszystkie niezbędne dla Ciebie usługi są w jednym miejscu!
Najczęściej unikamy zbędnych pytań Agencji w procesie rejestracji, jednak jeśli się pojawią, oferujemy wsparcie. Chętnie przejmiemy dyskusję z Agencją i zaproponujemy rozwiązania!
Jesteś zainteresowany przygotowaniem części klinicznej dokumentacji rejestracyjnej? A może kompleksową rejestracją produktu leczniczego? Skontaktuj się z nami!