

-
Rozwój produktu leczniczego
-
Badania kliniczne
-
Jakość
-
Audyty GxP
-
Usługi Rejestracyjne
-
Wsparcie przedrejestracyjne
- Regulatory roadmap
- Usługa Scientific Advice
-
Druki informacyjne
-
Dokumentacja rejestracyjna
- Audyt dossier
- Proces rejestracji
- Obsługa porejestracyjna
- Regulatory outsourcing
-
Wsparcie przedrejestracyjne
-
Pharmacovigilance
-
Usługa QP / Import
Wprowadzanie danych dotyczących produktów leczniczych – Zarządzanie bazą XEVMPD (baza z Artykułu 57)
Dlaczego posiadacze pozwoleń na dopuszczenie do obrotu muszą aktualizować dane produktu leczniczego w bazie XEVMPD?
Wszystkie firmy posiadające pozwolenia na dopuszczenie do obrotu produktów leczniczych na terenie Unii Europejskiej (UE) oraz Europejskiego Obszaru Gospodarczego (EOG) są prawnie zobowiązane do przekazywania Europejskiej Agencji Leków (EMA) informacji dotyczących zatwierdzonych produktów oraz do zapewnienia, że dane te pozostają aktualne. Obowiązek ten wynika z przepisów prawa farmaceutycznego Unii Europejskiej:
- Artykułu 57(2) Rozporządzenia (WE) nr 726/2004,
- Dyrektywy 2001/83/WE, zmienionej Dyrektywą 2010/84/UE
Europejska Agencja Leków opiera się na tych danych w celu wspierania procesów podejmowania decyzji regulacyjnych, analizy danych oraz działań komunikacyjnych.
Jaki jest cel przekazywania danych o produktach leczniczych do EMA?
Głównym celem przekazywania danych dotyczących produktów leczniczych jest stworzenie kompleksowej bazy danych obejmującej wszystkie produkty lecznicze dopuszczone do obrotu na terenie Unii Europejskiej (UE) oraz Europejskiego Obszaru Gospodarczego (EOG). Dotyczy to zarówno produktów dopuszczonych w procedurze centralnej za pośrednictwem Europejskiej Agencji Leków (EMA), jak i tych zatwierdzonych na poziomie krajowym.
Europejska Agencja Leków (EMA) wykorzystuje te dane w różnych celach, takich jak:
- Analiza danych i wykrywanie sygnałów,
- Zapewnienie zgodności z przepisami regulacyjnymi i prawnymi,
- Usprawnienie komunikacji z posiadaczami pozwoleń na dopuszczenie do obrotu.

Jakie obowiązki w zakresie przekazywania danych o produktach leczniczych spoczywają na posiadaczach pozwoleń na dopuszczenie do obrotu?
Przekazywanie danych dotyczących produktów leczniczych przez posiadaczy pozwoleń na dopuszczenie do obrotu jest obowiązkiem wprowadzonym w ramach przepisów dotyczących nadzoru nad bezpieczeństwem farmakoterapii (Dyrektywa 2010/84/UE oraz Rozporządzenie (UE) nr 1235/2010, Artykuł 57(2) Rozporządzenia (WE) nr 726/2004, Good Pharmacovigilance Practices (GVP) Moduł VI).
Każde nowe pozwolenie na dopuszczenie do obrotu, przyznane po 2 lipca 2012 r., musi zostać zgłoszone do Europejskiej Agencji Leków (EMA) w terminie 15 dni kalendarzowych od otrzymania powiadomienia do właściwego organu krajowego.
Wszelkie zmiany w pozwoleniu na dopuszczenie do obrotu – takie jak transfery pozwoleń, przedłużenia, zawieszenia, unieważnienia lub wycofania pozwoleń – muszą być zgłaszane do EMA nie później niż w ciągu 30 dni kalendarzowych od zatwierdzenia zmiany.
Dodatkowo, Europejska Agencja Leków (EMA) prowadzi migrację danych o produktach leczniczych z bazy xEVMPD do systemu Product Management Service (PMS), zgodnie ze standardami ISO IDMP, co ma na celu poprawę jakości danych. Proces ten wymaga od posiadaczy pozwoleń na dopuszczenie do obrotu standaryzacji informacji wprowadzonych do bazy xEVMPD.
Na czym polega nasza usługa Art. 57 / xEVMPD?
W SciencePharma świadczymy kompleksowe wsparcie w zakresie raportowania i aktualizacji danych produktów leczniczych w systemie xEVMPD – zarówno dla produktów już dopuszczonych do obrotu, jak i nowo rejestrowanych. Oferujemy pełna obsługę w przygotowaniu, weryfikacji i przesyłaniu danych – zapewniając zgodność, terminowość i spokój regulacyjny.
Nasze działania obejmują m.in.:
- Przygotowanie i rejestrację produktów w bazie xEVMPD,
- Weryfikację zgodności danych z wymaganiami EMA oraz IDMP,
- Aktualizację informacji i zgłaszanie zmian w wymaganym czasie (np. po zmianach w MA),
- Doradztwo przy migracji danych do systemu PMS (Product Management Service),
- Audyty jakości danych i przygotowanie zaleceń naprawczych.
Dla kogo jest ta usługa?
Nasza oferta skierowana jest do firm farmaceutycznych (MAHów), które posiadają pozwolenia na dopuszczenie do obrotu w krajach UE i EOG – niezależnie od tego, czy produkt został zatwierdzony w procedurze narodowej, MRP/DCP, czy centralnej.
Co zyskuje klient dzięki tej usłudze?
- Pewność zgodności z wymaganiami EMA oraz obowiązującym prawem UE – uniknięcie znalezisk w trakcje inspekcji z ramienia Agencji Rejestracyjnych. Znaleziska w trakcie inspekcji mogą prowadzić nawet do wycofania pozwolenia na dopuszczenie do obrotu – więcej informacji o inspekcjach i karach znajduje się tu,
- Odciążenie zespołów klienta i oszczędność czasu – my zajmujemy się całością procesu.
- Gotowość na przyszłe zmiany – np. migrację do systemu PMS
- Profesjonalne wsparcie techniczne i regulacyjne – zawsze chętnie odpowiemy na pojawiające się pytania,
- Redukcję błędów we wprowadzaniu danych do bazy – nasz proces zarządzania bazą xEVMP był wielokrotnie weryfikowany w trakcie audytów i inspekcji a członkowie działu PhV są regularnie szkoleni co zapewnia wysoką jakość naszych usług.
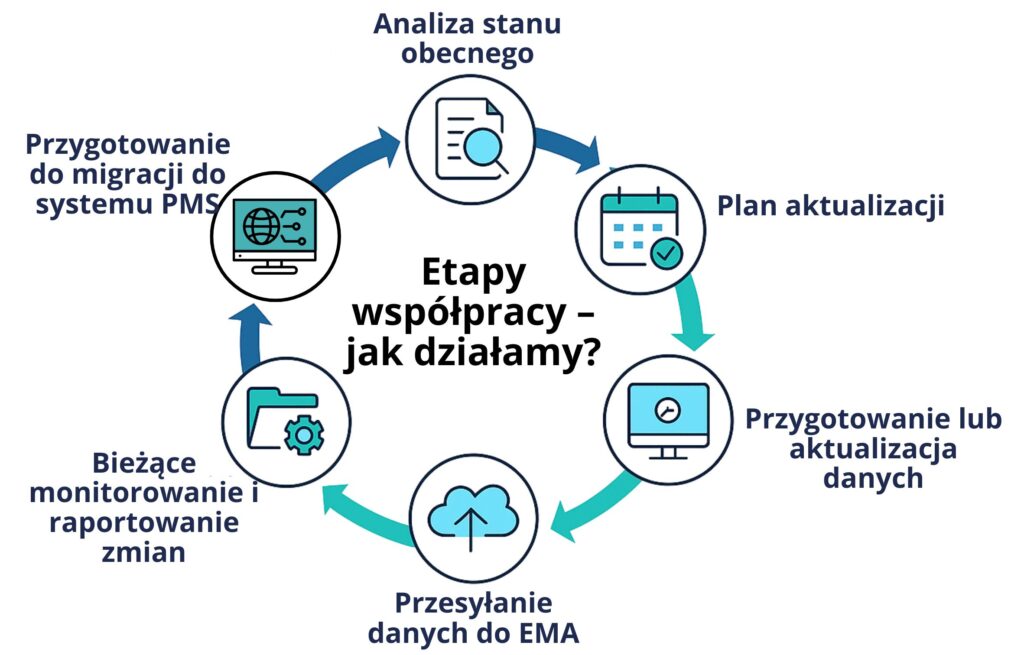
Etapy współpracy – jak działamy?
- Analiza stanu obecnego – weryfikacja kompletności danych w xEVMPD
- Plan aktualizacji – harmonogram działań i wymagane uzupełnienia
- Przygotowanie lub aktualizacja danych zgodnie z wymaganiami
- Przesyłanie danych do EMA – zgłoszenia w wymaganym formacie i potwierdzenie poprawności
- Bieżące monitorowanie i raportowanie zmian w cyklu życia produktu
- Przygotowanie do migracji do systemu PMS – standaryzacja danych i doradztwo

Dlaczego warto nas wybrać?
Dzięki wieloletniemu doświadczeniu oraz zespołowi zaangażowanych ekspertów zapewniamy kompleksowe wsparcie na każdym etapie cyklu życia produktu. Oferujemy nie tylko operacyjną obsługę, ale także strategiczne doradztwo, szkolenia lub audyty zgodności w tym zakresie.
Co nas wyróżnia?
- Udokumentowane doświadczenie – Nasza wieloletnia obecność na rynku mówi sama za siebie. Wykonaliśmy setki operacji w bazie xEVMPD zarówno dla produktów dopuszczonych centralnie, jak i krajowo. Posiadamy bogate doświadczenie w realizacji różnorodnych projektów. Współpracujemy z firmami różnej wielkości – od lokalnych podmiotów po globalnych liderów rynku.
- Zespół ekspertów – Nasi specjaliści posiadają wieloletnie doświadczenie w pracy z bazą xEVMPD i doskonałą znajomość regulacji EMA, w tym standardów ISO IDMP i wytycznych GVP. Regularnie uczestniczą w szkoleniach i śledzą zmiany w przepisach, dzięki czemu zapewniają najwyższy poziom merytoryczny obsługi.
- Aktualne procesy – Nieustannie dostosowujemy nasze procedury do najnowszych standardów branżowych oraz wymagań regulacyjnych. Pracujemy według zaktualizowanych procedur zgodnych z wymaganiami EMA i zapewniamy pełną audytowalność działań.
- Kompleksowa obsługa cyklu życia produktu – Oferujemy pełne wsparcie we wszystkich procesach związanych z cyklem życia produktu. Obsługujemy cały proces zarządzania danymi w xEVMPD: od zgłoszenia nowego produktu, przez aktualizacje po zmianach w pozwoleniu, aż po wsparcie przy migracji danych do systemu PMS. Działamy proaktywnie, monitorując zmiany statusów pozwoleń i przypominając o obowiązkach terminowych.
Nawiąż współpracę z nami i miej pewność, że Twój produkt znajduje się w rękach ekspertów.
SciencePharma zapewnia profesjonalne wsparcie w zakresie pharmacovigilance przez cały cykl życia produktu leczniczego. Nasze usługi są starannie dopasowane do indywidualnych wymagań każdego klienta. Dzięki wdrożeniu zaawansowanego systemu zarządzania bezpieczeństwem leków, umożliwiamy naszym klientom skoncentrowanie się na strategicznym rozwoju ich produktów, jednocześnie zachowując najwyższe standardy bezpieczeństwa pacjentów oraz zgodności z przepisami regulacyjnymi.
Skontaktuj się z nami i dowiedz się, jak możemy wesprzeć Twoją organizację w zapewnieniu pełnej zgodności regulacyjnej i bezpieczeństwa farmakoterapii.
Źródła:
- https://www.ema.europa.eu/en/documents/other/data-submission-authorised-medicines-european-union-outlines-article-572-regulation-ec-no-7262004_en.pdf
- https://www.ema.europa.eu/en/data-medicines-iso-idmp-standards-post-authorisation/extended-eudravigilance-medicinal-product-dictionary-xevmpd-training
- https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/data-medicines-iso-idmp-standards-post-authorisation/data-submission-authorised-medicines-article-57